Cell culturing and seeding of cells. TIMING 3-7 days depending on the cell line
1. Grow low-passage epithelial cells to 70-80% confluence in 75 cm2 tissue culture flasks. Ca9-22 and HaCat cells should be grown in DMEM supplemented with 10% FCS, 100 U/ml penicillin and 100 μg/ml streptomycin. HO-1-N-1 cells should be cultured in DMEM/F12 (1:1) supplemented with 10% FCS, 100 U/ml penicillin and 100 μg/ml streptomycin.
CRITICAL STEP. Always wear gloves when handling cells and work in a laminar flow cabinet. The cell culture environment should be kept clean, i.e. in absence of bacteria, molds and mycoplasma.
2. Discard the growth culture media and wash the cells with 6 ml PBS.
3. Discard the PBS and add 2 ml trypsin/EDTA and incubate at 37ºC for 3-5 minutes.
CRITICAL STEP. Verify complete detachment of the cells under an inverted microscope.
4. Add 8 ml of supplemented growth media into the flask and gently resuspend the cells by gently pipetting up and down.
5. Take an aliquot of the cell suspension and determine the cell counts using a counting chamber. Dilute the cell suspensions in the corresponding growth media. The Ca9-22 cell suspension should be diluted to obtain 3.5 x 105 cells/ml; whereas HO-1-N-1 and HaCat cell suspensions should be diluted to obtain 3.0 x 105 cells/ml.
CRITICAL STEP. The number of cells required to form an intact monolayer within the standardized timeframe depends on the cell type and thus, should be pre-determined for each cell line.
6. Mix the cell suspension gently and add the suspension into a reagent reservoir.
7. Using a multichannel pipette, add 100 µl of the cell suspension into each well of a 96-well plate with flat bottom (BD Falcon; recommended for imaging using the BD Pathway 855 Bioimaging System).
8. Incubate for 16 hours in a humidified incubator at 37ºC with 5% CO2.
Cell starvation. TIMING 2 hours (might vary for other cell lines)
9. Replace the growth media by 100 µl starvation media (i.e. FCS-free media) by aspiration of the culture medium without disturbing the cell monolayers. HaCat cells should be starved in keratinocyte serum-free medium (SFM). Starvation of Ca9-22 cells should be performed in FCS-free DMEM/F12, whereas HO-1-N-1 cells should be starved in FCS-free DMEM. SFM is not recommended when using Ca9-22 or HO-1-N-1 cells as it was shown to promote significant cell migration and proliferation in our setup that restricted the detection of exogenously added stimulatory compounds.
CRITICAL STEP. The duration and the media used during starvation need to be optimized for each cell line. FCS deprivation should minimize cell migration and proliferation without inducing apoptosis or cell detachment. ? TROUBLESHOOTING
10. Incubate the cells (Ca9-22, HO-1-N-1 or HaCat cells) for 2 hours in a humidified incubator at 37ºC with 5% CO2.
Preparation of master plate with treatment and control samples. TIMING 1.5 hours
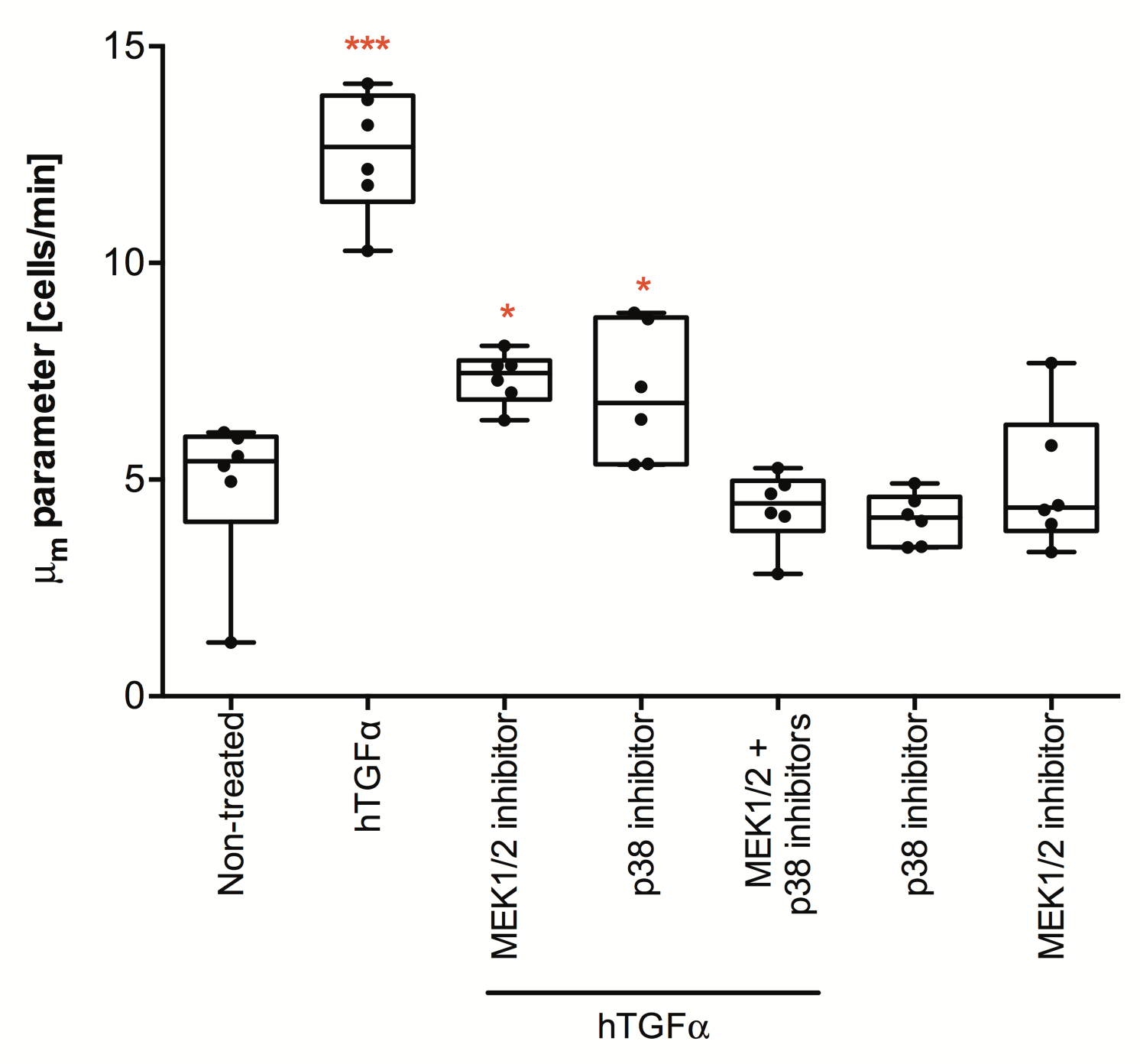
11. Prepare the treatments that you wish to test for their effect on re-epithelialization kinetics. An example of bacterial preparations is provided in Figure 2. Initial screenings can be performed in duplicates, but refined studies should be carried out with at least three technical replicates in two or more independent experiments.
CRITICAL STEP. All treatments should be diluted in the starvation media.
12. Prepare the positive and negative controls as indicated in Figure 3.
CRITICAL STEP. Optimal concentrations of the controls should be determined by performing a titration for the target cell lines (Supplementary Fig. 1) and should result in a large dynamic range between the positive and negative controls (Figure 1). ? TROUBLESHOOTING
13. Prepare a master plate by adding 120 μl of each sample into the corresponding well.
CRITICAL STEP. Exclude outer wells to avoid any possible edge-specific artifacts, e.g. due to evaporation of the medium. It is also recommended to add the samples into the wells in a randomized manner.
Fluorescent labelling with live-compatible dyes. TIMING 25 minutes
14. Prepare a solution of starvation media with the fluorescent dyes. Add 10 μl of Hoechst 33342 working solution and optionally 100 μl of CellTracker™ Red CMTPX stock solution into 5 ml starvation media to obtain a final concentration of 2 μg/ml and 2 μM, respectively.
CRITICAL STEP. Nuclear labelling with Hoechst 33342 stain is crucial for accurate image segmentation and feature extraction with the automated image analysis pipeline presented in this protocol. Labelling of cellular cytoplasm with CellTracker™ Red CMTPX can be performed to visualize cell boundaries. The concentrations of these dyes may need to be adjusted depending on the selected cell line. When using a different cell line, check that addition of the dyes at the desired concentration does not result in adverse effects in re-epithelialization kinetics. ? TROUBLESHOOTING
15. Add the labelling solution into a reagent reservoir and carefully aspirate the starvation media from the cells using a vacuum pump without disturbing the cell monolayer.
16. Add 50 μl of the solution to each of the wells and incubate at 37ºC with 5% CO2 for 20 minutes.
Scratch assay and imaging. TIMING 5-16 hours depending on the cell line
17. After 20 minutes of incubation with the labelling solution, perform a scratch in the cell monolayers of each well using the HTSScratcher to assure equally sized and reproducible scratches. Place the plate on the platform, slide the plate under the 96-pin array, press the array towards the bottom of the wells, and slide the array from left to right 3 times. Release pressure from the array and remove the plate from the platform.
CRITICAL STEP. Rinse the array with water followed by ethanol before and after each experiment. ? TROUBLESHOOTING
18. Carefully aspirate the liquid from each well using the vacuum pump and wash the cells with 100 µl PBS. Repeat this step one more time.
19. Aspirate the PBS out and immediately transfer 100 µl of each sample from the master plate to the corresponding wells in the assay plate using a multichannel pipette.
CRITICAL STEP. Add 100-200 µl PBS in the outer wells to avoid differential evaporation of the samples. ? TROUBLESHOOTING
20. Start image acquisition. For the Ca9-22 or HO-1-N-1 cell lines, we recommend programming the microscope to acquire images every 20 minutes for a total of 5 hours. For the HaCat cell line, imaging intervals can be adjusted to 60 minutes for a total of 14-16 hours.
CRITICAL STEP. The duration of the assay may vary depending on the cell line and can be determined by the time required for the positive control to achieve wound closure.
Image and data analysis. TIMING 45 minutes
21. For first time users, download the virtual machine (VM) containing the Kinetic Re-Epithelialization Analysis Pipeline (KREAP) toolbox as well as the VM player for your operating system via https://erasmusmc-bioinformatics.github.io/KREAP/. Import the VM into the player following the step-by-step instructions provided in the manual on the website.
22. Create an index file (.txt) and a compressed plate folder (.zip) containing the acquired images organized in separate folders for each well. Indications on how to create these files are provided at https://erasmusmc-bioinformatics.github.io/KREAP/file_formats.
23. Upload the compressed folder and index file into the Galaxy history via the “Get data” tool.
CRITICAL STEP. Set the compressed folder’s type to .zip in the upload menu.
24. Run the KREAP Image Analysis tool to perform automated image segmentation. The results are reported in an HTML format and can be downloaded and stored locally. A manual is provided at https://erasmusmc-bioinformatics.github.io/KREAP/use_kreap_analysis. ? TROUBLESHOOTING
25. Run the KREAP Data-Modeling tool to quantify re-epithelialization kinetics. The results are reported in an HTML format and can be downloaded and stored locally. A manual is provided at https://erasmusmc-bioinformatics.github.io/KREAP/use_kreap_modeling.
CRITICAL STEP. Remember to set the time interval \(min) between images in the index file. ? TROUBLESHOOTING
{kind=link}