Step1-3:
Preparation and culture of ES-derived or iPS-derived neurons
- Freeze the neurons:
1.1) Check the culture under the microscope to confirm that the neurons are viable and have the correct morphology (Fig. 3a).
1.2) Treat the culture with the caspase inhibitor z-VAD-FMK at 500 nM for 30 minutes in the incubator by adding the inhibitor directly to the culture medium;
1.3) In the tissue culture hood, aspirate the culture medium from the dishes, and then add 1XDPBS into the dish to wash out the residue medium; ~2 ml per 35 mm Petri dish, or proportional volumes for other sizes of culture surfaces;
1.4) Aspirate the DPBS and add pre-warmed Accutase to lift the cells; ~0.5 ml per 35 mm Petri dish, or proportional volumes for other sizes of culture surfaces;
CAUTIONS: Be quick at all of the above steps so that the culture does not get dry for over ~5 seconds.
1.5) Put the dishes into the 37oC tissue culture incubator, and incubate them for ~15 minutes;
1.6) Check the culture under the microscope to confirm that the neurons round up and about to detach the dish (Fig. 3b). If the cells have not reached this type of morphology, put the dishes back into the incubator and check every ~3 minutes again until the cells are about to detach.
★CRITICAL STEP: The cells should be about to detach in at most 30 minutes; do not incubate the cells in Accutase for more than 30 minutes.
1.7) Gently scrap the neurons off the dish with a cell scrapper and dissociate possible clumps via gentle pipetting for 3~5 times so that the neurons are lifted into single suspended cells (Fig. 3c).
★CRITICAL STEP: Be slow and gentle during scraping and pipetting so that the neurons are not damaged.
1.8) Add pre-warmed culture medium A (see REAGENT SETUP) into 15 ml or 50 ml conical tubes and transfer the cells into it. The volume of the added culture medium is 9 times of the Accutase volume utilized to lift the neurons.
1.9) Spin the cells at 1000 rpm for 4 minutes to collect the cells; save a small aliquot for cell counting and viability measurement.
★CRITICAL STEP: Do not spin at much higher speed to avoid possible damage of the cells and possible precipitation of death cells or cell debris. Cell counting and viability measurement could be performed by loading the saved cell suspension onto Vi-Cell XR Cell Viability Analyzer for automatic analysis or by standard Trypan-blue staining and counting on hemocytometers.
1.10) During spinning, count the cell density and viability based on Trypan-blue exclusion; the viability needs to be close or above 90%.
1.11) Aspirate the medium from the conical tubes and resuspend the cells in freezing medium; calculate the volume of freezing medium so that cell density is about 5~10 X 106/ml.
1.11) Aliquot the cells into cryopreservation tubes at 1ml per vial.
1.12) Put the vials into the cell freezing container, and then keep the boxes into -80 oC freezer for one or more days; transfer the vials into cyropreservation boxes into liquid N2 tanks for long term storage.
★CRITICAL STEP: Nalgene Cryo freezing container or similar container should be used to achieve a −1°C per minute rate of cooling when placed at −80°C.
- Coat the dishes (Day -1):
2.1) In the tissue culture hood, pre-cool the dishes in a clean, ethanol sprayed metal tray on ice.
2.2) Pre-cool a tube of X-Vivo 10 medium on ice.
2.3) Pipette up and down the cooled X-Vivo 10 medium to cool down the 5 ml pipette tube.
2.4) Prepare 1:30 matrigel by mixing 1 volume of matrigel with 29 volumes of X-Vivo 10 medium on ice via pipetting; keep the diluted matrigel and the culture dishes cold.
★CRITICAL STEP: pre-cool and keep everything cold so that matrigel does not precipitate; the pipette tips could be cooled by pipetting pre-cooled X-Vivo 10 medium.
2.5) Coat the culture dishes by adding the diluted matrigel into each dish, 2 ml per 35 mm Petri dish, or proportional volume for dishes with other sizes of culture surface; transfer the dishes into the 4 oC fridge and coat overnight.
PAUSE POINT: Keep the dishes in 4 oC fridge overnight.
CAUTION1: Coat the dishes at least one day before starting a culture.
CAUTION2: The coated dishes should be used within a week, and should not be used if stored for over a week or left at above room temperature for over a day.
CAUTION3: Use a single lot of matrigel for a batch of culture to avoid inconsistencies that may be created by different lots.
- Start a culture:
3.1) Warm up the coated dishes in 37 oC incubator for ~30 minutes.
★CRITICAL STEP: Check the dishes under microscope to confirm that the coating is successful (Fig. 3d). Do not remove the matrigel medium until the time point for plating to avoid complete dryness of the plate.
Option (A): for ESC derived neurons
i) Pre-warm complete culture medium A (see REAGENT SETUP) in 37 oC bath.
ii) Thaw the cells by incubating the frozen vials in 37 oC bath for ~2 minutes with gentle shaking until most content becomes liquidized.
iii) Briefly mix the content in the vial by gentle pipetting, and then transfer the content into the pre-warmed culture medium A (9 ml for each thawed vial) in conical tubes.
iv) Collect the thawed cells by spinning at 1000 rpm for 4 minutes; save an aliquot for measuring cell density and viability.
v) During spinning, count the cell density and viability based on Trypan-blue exclusion; the viability needs to be close or above 70%.
vi) Aspirate the medium and add culture medium A to resuspend the cells to achieve a concentration of 1X106 cells/ml.
vii) Aspirate the matrigel and plate the cells to coated culture dishes at 2 ml per 35 mm Petri dish, or proportional volume for dishes with other sizes of culture surfaces.
Caution: Add the cells to the dishes immediately after aspirating the matrigel so that the coated matrix does not get completely dry.
viii) Change the medium the next day with fresh pre-warmed complete culture medium; check the culture to ensure that the neurons look fine (Fig. 3e).
★CRITICAL STEP: Change culture medium with fresh ones every three days. The thawed cells should be used for subsequent steps within a week to avoid possible mild neuronal death of disease neurons over very long term even under protective conditions.
Option (B): for iPSC derived neurons
The procedure is the same as Option (A) in this step, except that culture medium B (see REAGENT SETUP) is used instead of culture medium A.
Step 4-5:
siRNA tranfection of ESC-derived or iPSC-derived neurons
- Transfection mix preparation
4.1) dissolve the purchased siRNA with OPTI-MEM to reach a concentration at 10 μM.
★CRITICAL STEP: For purchased siRNA libraries, the siRNA are normally stamped at the bottom of 96-well plates. Add the water gradually around the bottom of each well and then pipette up and down slowly with multi-channel pipettes to dissolve the stamped siRNA completely. Spin down at >1000 rpm for 1 minutes to collect all the dissovled siRNAs.
4.2) Transfer 3 μl of each siRNA to each well of the 96-well clear bottom PDL-coated plates for Option (A)&(B), or matrigel-coated plates for Option (C)&(D), by multi-channel pipettes; the options are listed in step 5 shown as below.
4.3) Add 1 volume of Lipofectamine RNAiMax to 9 volumes of Opti-MEM and mix by inverting the tubes for ~5 times and then incubate that room temperature for 5 minutes.
4.4) Add 3 μl of Lipofectamine RNAiMax containing Opti-MEM into the siRNA in each well by multi-channel pipettes and mix by gentle pipetting for ~ 3 times.
4.5) Put the lids back on the plates, and incubate the transfection mix in the hood for 30 minutes; incubation up to 1 hour is fine.
- Plating the cells:
5.1) While incubating the transfection mix, lift the ESC or iPSC derived neurons via the same procedure as described in step1.3 through 1.10
★CRITICAL STEP: Most of the dead cells after thawing should have been discarded already during medium change and cell collection procedure above. The remaining cell viability of the collected cells needs to be close to or above 90% for the following steps.
Option (A): for neurodegeneration measurements of ESC-derived neurons
i) Resuspend the cell pellet with complete culture medium C to reach a concentration of 1.8~2 X 106 cells/ml; pipette up and down gently to mix the cells.
CAUTIONS: Pipette slowly and gently to avoid air bubbles and possible damages to the cells; no clumps of cells should be visible in the tube after mixing.
ii) Add 54 μl of the cell suspension on top of the transfection mix in each well of the PDL-coated 96-well plates; avoid the air bubbles when pipetting.
iii) Mix the cells with the transfection mix by horizontal shaking of the plates.
iv) Put the plates into the 37oC tissue culture incubator for 6 hours.
v) Add 180 μl of pre-warmed complete culture medium C in each well by multi-channel pipettes.
Option (B): for neurodegeneration measurements of iPSC-derived neurons
The procedure is the same as Option (A) in this step, except that culture medium D (see REAGENT SETUP) is used instead of culture medium C.
Option (C): for siRNA knock-down experiments under protective condition in ESC-derived neurons (use this option for protein measurements in these neurons)
The procedure is the same as Option (A) in this step, except that culture medium A is used instead of culture medium C, and matrigel-coated plates are used instead of PDL-coated plates.
Option (D): for siRNA knock-down experiments under protective condition in iPSC-derived neurons (use this option for protein measurements in these neurons)
The procedure is the same as Option (A) in this step, except that culture medium B is used instead of culture medium C and matrigel-coated plates are used instead of PDL-coated plates.
Step 6-8:
Neurodegeneration Measurement
For step 5 Option (A) and (B) plates only, place each plate into the IncuCyte inside the incubator (Fig. 1)
★CRITICAL STEP: Ensure that the underside of each plate is clean and free from dust. If required, a lint free tissue moistened with 70% ethanol can be used to carefully wipe the underside. After placing the plates into the IncuCyte, ensure that each plate is fully inserted into the microplate tray holder; a maximum of six plates can be loaded at a time.
IncuCyte setup for data collection
Option (A): for confluence measurement based phase-contrast images
i) In the IncuCyte software, select the “Schedule Upcoming Scans” option and populate the gantry view with the plate information (Fig. 2a-c). For each plate select the appropriate tray location and choose Tray Type: Microplates. Under Vessel Type select the appropriate definition for the microplate used.
★CRITICAL STEP It is important that the correct vessel definition is chosen to ensure optimal imaging. Over 140 different 96-well plates are supported for use with IncuCyte. The correct vessel definition can be found by referring to the catalogue number for the plate. For these experiments a BD Falcon Optilux plate was used.
ii) Under “Scan Type” select “Phase-Contrast”
iii) Select “Edit Scan Patterns” and create a new pattern by selecting all 96 wells and 4 fields per well for image collection.
★CRITICAL STEP Apply the newly created scan pattern to each plate. The scan pattern need only be created once after which it will be available for use with future experiments (Fig. 2a-c).
iv) Under the “Properties” tab add an appropriate Label for the plate and provide information about the cells used (Fig. 2b).
v) Right-click on the “Timeline” to set a new scan interval; schedule 24 hour repeating scans every 4 to 12 hours (Fig. 2a&e).
CAUTIONS: Choose an appropriate scan frequency. IncuCyte will slowly generate heat when actively scanning. Excessive scanning can cause a temperature increase within the incubator. It is recommended that no more than 12 hours of active scanning is scheduled every 24h and that a continuous scan time of 45 minutes is not exceeded. IncuCyte will warn the user if these recommendations are exceeded and cooling periods (Fig. 2d) can be used to maximize the duty cycle without causing temperature increase. In contrast, insufficient scanning can lead to poorly defined kinetics. The recommended scan frequency of 4 to 12 hours is a good balance.
★CRITICAL STEP Let the plates equilibrate within the IncuCyte for at least 10 minutes before commencing the first scan. This will ensure that any condensation present on the underside of the plates has evaporated prior to imaging.
vi) Click the “Apply” button to initiate the changes to the scan schedule, and the scan will start at the next scheduled time point after the current time.
Option (B): for caspase-3 activity measurement based fluorescent images
i) Perform a 1:10 dilution of the NucView 488 caspase-3 dye with Opti-MEM
ii) Add 2 µl of the diluted dye into each well 24 hours after siRNA transfection in iPSC-derived neurons.
iii) Setup the IncuCyte software the same way as Option (A) i) through vi), except that under the “Scan Type” select “Fluorescence and Phase-Contrast”; the fluorescent images taken 24 hours after adding the dye could then be used for analysis.
- Data collection and analysis
Option (A): Confluence measurements from phase-contrast images
i) During the course of the assay review the high definition phase contrast images to ensure assay quality (Fig. 4a). Plot the kinetic “Confluence v1.5” data regularly with the IncuCyte software to get a real-time overview of treatment effects and to determine a suitable point to end the assay (usually 3 to 4 days). Assay length will depend on the neuronal model used and the experimental design. Export the “Confluence v1.5” data using the IncuCyte software for analysis (Fig. 2e).
CAUTIONS: if the experiments requires 7 days or longer, change ~75% of the culture medium with fresh ones every 7 days.
ii) Plot the Confluence v1.5 metric for each condition over time (Fig. 5b-c)
iii) Establish threshold criteria to determine if certain modifiers show rescue effects (Fig. 5b-c)
Option (B): for caspase-3 activity measurement based fluorescent images
i) 24 hours after adding the dye, review the phase contrast and green channel fluorescence images to ensure assay quality (Fig. 4b).
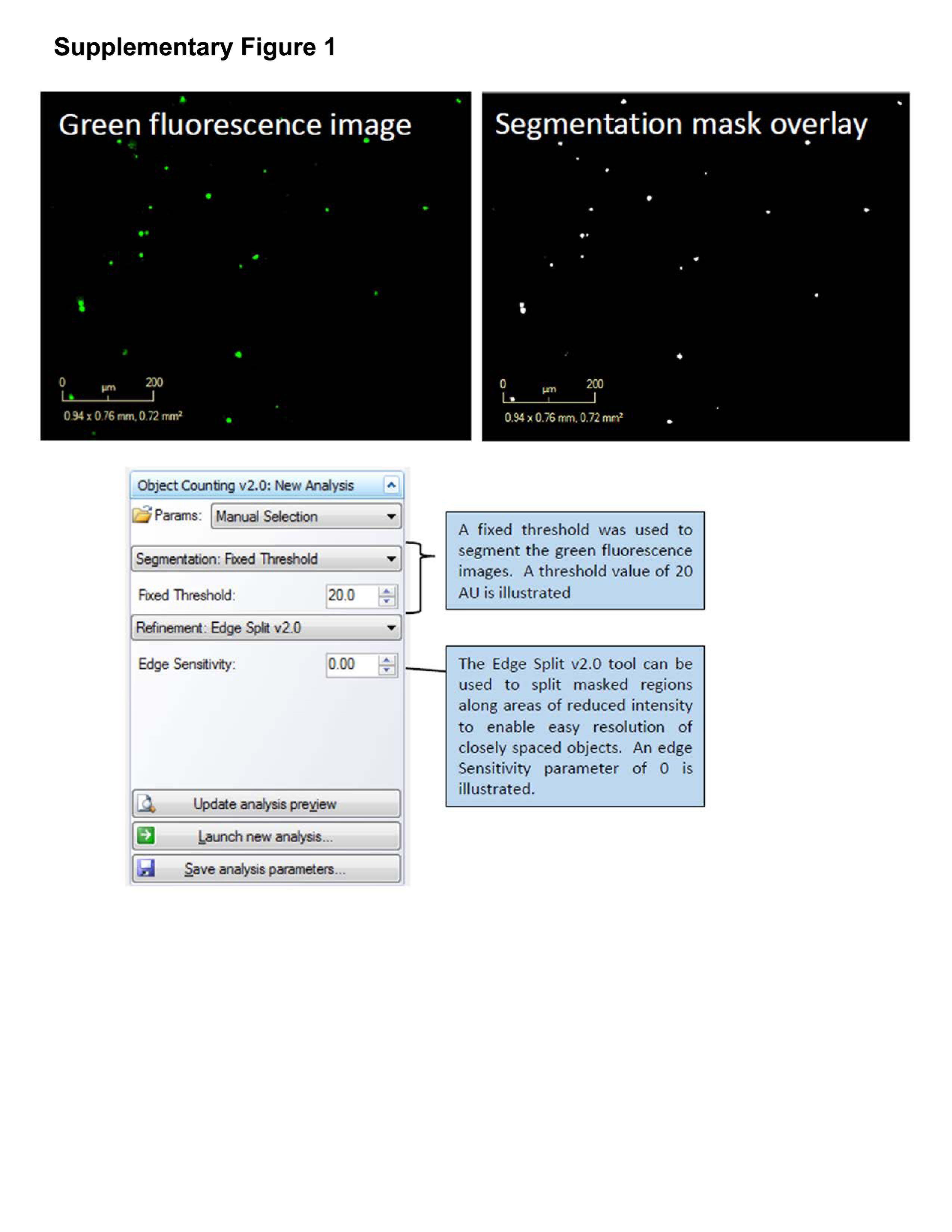
ii) Within the IncuCyte software analyze the fluorescence images using the integrated “Object Counting v2.0 algorithm”; choose Segmentation: Fixed Threshold and input a suitable threshold value to differentiate foreground fluorescent cells (caspase-3/7 active objects) from background fluorescence (Supplementary Fig. 1). The Edge Split v2.0 tool can be used to separate adjacent objects that would otherwise be masked together and filters can be applied (size, shape and intensity) to further refine image segmentation.
iii) Review the fluorescence image analysis by plotting the “Object Confluence metric”. This fluorescence channel metric represents the percentage of the total field of view occupied by masked foreground objects. Ensure that the metric is consistent with the fluorescence image data and that is accurately reflects the neurodegenerative phenotypes observed.
iv) Export the “Object Confluence” and “Confluence v1.5” metrics (Fig. 2e)
v) Normalize the kinetic “Object Confluence” data to the initial “Confluence v1.5” value; plot the bar graph of the normalized caspase-3 signals8.
{kind=link}